PEG
![]()
PTHF
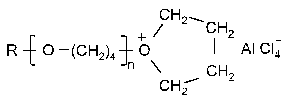
PCLT
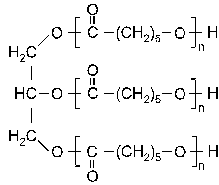
HDI
![]()
TMP
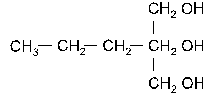
DD
![]()
| Polyether | ||
| Polyethylenglykol |
PEG |
|
| Polytetrahydrofuran |
PTHF |
|
| Polyester | ||
| Poly(caprolacton)triol |
PCLT |
|
| Isocyanat | ||
| 1,6-Hexamethylendiisocyanat |
HDI |
|
| Polyole | ||
| 1,1,1-Tris(hydroxymethyl)propan |
TMP |
|
| 1,10-Decandiol |
DD |
|
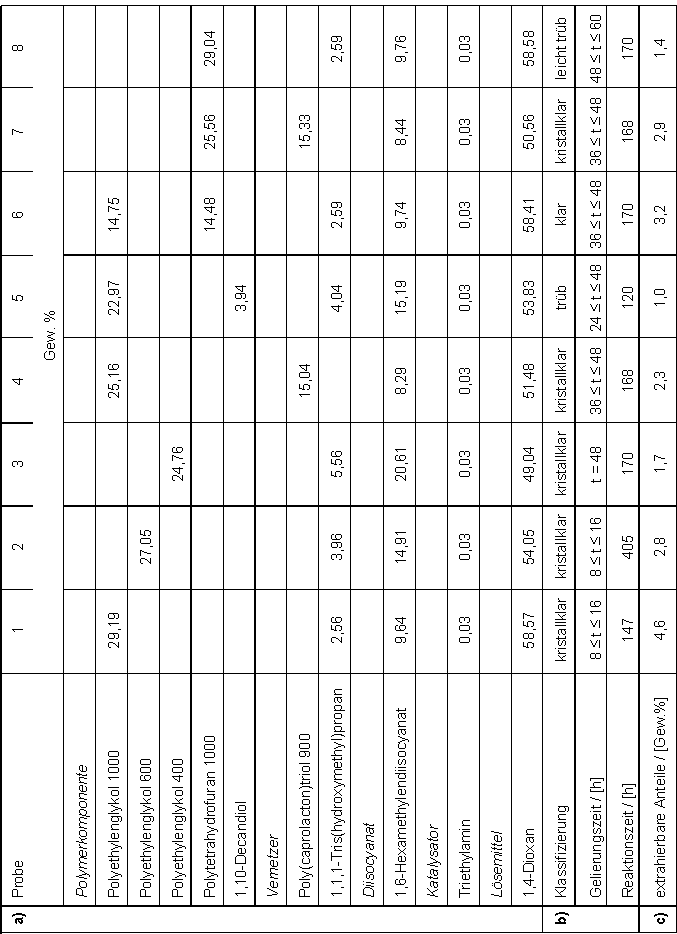
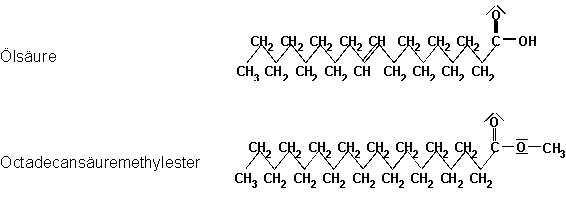 Abb.(6.1): Darstellung der Strukturformel der als Quellmittel verwendeten flüssig-kristallinen Substanzen.
Aus Vorversuchen hat sich ergeben, daß lediglich drei der Polyurethan-Netzwerke, die Proben 3, 6 und 8, für die Quellungsexperimente in Ölsäure bzw. Octadecansäuremethylester geeignet sind. Mit leichten Einschränkungen konnten für diese Netzwerke reproduzierbare Quellungsgrade bestimmt werden. Bei der Durchführung der Probenwägungen zeigte sich, daß aufgrund der öligen Konsistenz der beiden Quellmittel, die Netzwerkproben nur schwer vom anhaftenden Quellmittel befreit werden können. Ein Herauslösen der Netzwerkprobe aus dem Quellmittel wird bei dem Octadecansäuremethylester zusätzlich dadurch erschwert, daß dieses Quellmittel bei Raumtemperatur kristallin vorliegt. Bei der Entfernung des Octadecansäuremethylesters von der Netzwerkprobe konnte zum Teil eine Beschädigung des Netzwerks nicht verhindert werden. Deshalb sind in diesem Fall nur für die Netzwerke 3 und 8 reproduzierbare Ergebnisse erzielt worden. Bei den Experimenten mit Ölsäure zeigte sich, daß diese möglicherweise die Netzwerkbindungen der Polyurethangele zersetzt. Hier stehen daher nur Daten für die Netzwerke 3 und 6 zur Verfügung.
In zeitabhängig durchgeführten Quellungsexperimenten, die in Abb.(6.2) und (6.3) dargestellt sind, wurde die Zeitspanne bis zur Konstanz des Quellungsgrades festgestellt. Es wird davon ausgegangen, daß dann das Quellungsgleichgewicht vorliegt. Da bei den untersuchten Systemen kleine Diffusionskoeffizienten vorliegen, wurden lange Einstellzeiten beobachtet. Für die Quellung in Ölsäure muß eine Zeitspanne von etwa 25 Tagen, bei der Quellung in Octadecansäuremethylester von 20 Tagen bis zur Einstellung des Quellungsgleichgewichtes abgewartet werden.
Abb.(6.1): Darstellung der Strukturformel der als Quellmittel verwendeten flüssig-kristallinen Substanzen.
Aus Vorversuchen hat sich ergeben, daß lediglich drei der Polyurethan-Netzwerke, die Proben 3, 6 und 8, für die Quellungsexperimente in Ölsäure bzw. Octadecansäuremethylester geeignet sind. Mit leichten Einschränkungen konnten für diese Netzwerke reproduzierbare Quellungsgrade bestimmt werden. Bei der Durchführung der Probenwägungen zeigte sich, daß aufgrund der öligen Konsistenz der beiden Quellmittel, die Netzwerkproben nur schwer vom anhaftenden Quellmittel befreit werden können. Ein Herauslösen der Netzwerkprobe aus dem Quellmittel wird bei dem Octadecansäuremethylester zusätzlich dadurch erschwert, daß dieses Quellmittel bei Raumtemperatur kristallin vorliegt. Bei der Entfernung des Octadecansäuremethylesters von der Netzwerkprobe konnte zum Teil eine Beschädigung des Netzwerks nicht verhindert werden. Deshalb sind in diesem Fall nur für die Netzwerke 3 und 8 reproduzierbare Ergebnisse erzielt worden. Bei den Experimenten mit Ölsäure zeigte sich, daß diese möglicherweise die Netzwerkbindungen der Polyurethangele zersetzt. Hier stehen daher nur Daten für die Netzwerke 3 und 6 zur Verfügung.
In zeitabhängig durchgeführten Quellungsexperimenten, die in Abb.(6.2) und (6.3) dargestellt sind, wurde die Zeitspanne bis zur Konstanz des Quellungsgrades festgestellt. Es wird davon ausgegangen, daß dann das Quellungsgleichgewicht vorliegt. Da bei den untersuchten Systemen kleine Diffusionskoeffizienten vorliegen, wurden lange Einstellzeiten beobachtet. Für die Quellung in Ölsäure muß eine Zeitspanne von etwa 25 Tagen, bei der Quellung in Octadecansäuremethylester von 20 Tagen bis zur Einstellung des Quellungsgleichgewichtes abgewartet werden.
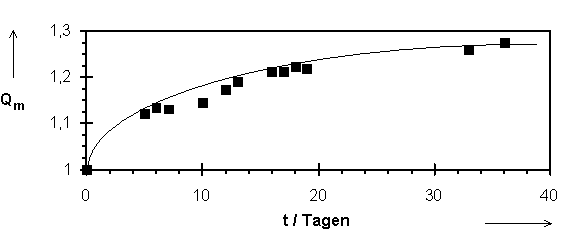 Abb.(6.2): Darstellung der Zeitabhängigkeit der Einstellung des Quellungsgleichgewichts für das System Netzwerk 6/Ölsäure bei 45°C.
Abb.(6.2): Darstellung der Zeitabhängigkeit der Einstellung des Quellungsgleichgewichts für das System Netzwerk 6/Ölsäure bei 45°C.
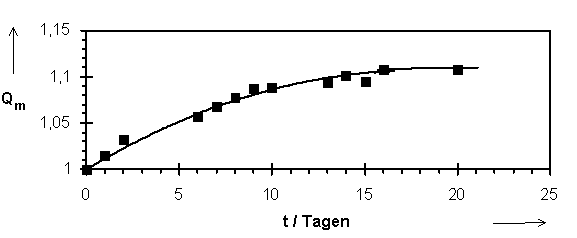 Abb.(6.3): Darstellung der Zeitabhängigkeit der Einstellung des Quellungsgleichgewichts für das System Probe 3/Octadecansäuremethylester bei 45°C.
Abb.(6.3): Darstellung der Zeitabhängigkeit der Einstellung des Quellungsgleichgewichts für das System Probe 3/Octadecansäuremethylester bei 45°C.