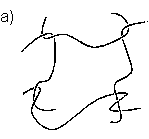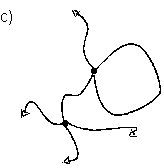|
|
|
|
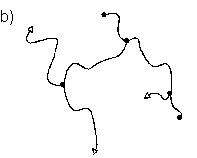
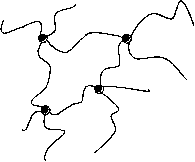 Abb.(2.1): Zweidimensionaler, schematischer Ausschnitt eines tetrafunktionalen, chemisch verknüpften Netzwerks.
Abb.(2.1) zeigt einen schematischen, zweidimensionalen Ausschnitt eines solchen Netzwerks. Die Knotenpunkte des gezeigten Netzes sind tetrafunktionell. Damit ist gemeint, daß von jedem Knotenpunkt vier Polymerketten ausgehen, die wiederum zu Knotenpunkten der Funktionalität f=4 führen.
Allgemein müssen die Verknüpfungsstellen eines Netzwerks, unabhänigig von der Art der Verknüpfung, eine mittlere Funktionalität f > 2 aufweisen, damit eine Vernetzung der Polymerketten vorliegt. Liegt die Funktionalität der Knotenpunkte bei f = 2, werden in einer Verknüpfungsreaktion lineare Ketten gebildet. Kennzeichen chemisch verknüpfter Netzwerke ist ihre Stabilität gegenüber Lösemittel und Temperatur, weshalb sie auch als "irreversible" Netzwerke bezeichnet werden. Bei erhöhten Temperaturen zeigen solche Netzwerke öfter das Verhalten weicher aber elastischer Feststoffe als das viskoser Flüssigkeiten [6]. Hierfür ist vulkanisierter Kautschuk das bekannteste Beispiel.
b) Physikalisch vernetzte Polymernetzwerke:
Bei den über physikalische Bindungen verknüpften Netzwerken handelt es sich im allgemeinen um sogenannte thermoreversible Gele. In Abhängigkeit von der Temperatur oder auch der Menge zugefügtem Lösemittel erfolgt bei diesen Polymer/Lösemittel Systemen ein reversibler Gel-Sol-Übergang [7-11].
Abb.(2.1): Zweidimensionaler, schematischer Ausschnitt eines tetrafunktionalen, chemisch verknüpften Netzwerks.
Abb.(2.1) zeigt einen schematischen, zweidimensionalen Ausschnitt eines solchen Netzwerks. Die Knotenpunkte des gezeigten Netzes sind tetrafunktionell. Damit ist gemeint, daß von jedem Knotenpunkt vier Polymerketten ausgehen, die wiederum zu Knotenpunkten der Funktionalität f=4 führen.
Allgemein müssen die Verknüpfungsstellen eines Netzwerks, unabhänigig von der Art der Verknüpfung, eine mittlere Funktionalität f > 2 aufweisen, damit eine Vernetzung der Polymerketten vorliegt. Liegt die Funktionalität der Knotenpunkte bei f = 2, werden in einer Verknüpfungsreaktion lineare Ketten gebildet. Kennzeichen chemisch verknüpfter Netzwerke ist ihre Stabilität gegenüber Lösemittel und Temperatur, weshalb sie auch als "irreversible" Netzwerke bezeichnet werden. Bei erhöhten Temperaturen zeigen solche Netzwerke öfter das Verhalten weicher aber elastischer Feststoffe als das viskoser Flüssigkeiten [6]. Hierfür ist vulkanisierter Kautschuk das bekannteste Beispiel.
b) Physikalisch vernetzte Polymernetzwerke:
Bei den über physikalische Bindungen verknüpften Netzwerken handelt es sich im allgemeinen um sogenannte thermoreversible Gele. In Abhängigkeit von der Temperatur oder auch der Menge zugefügtem Lösemittel erfolgt bei diesen Polymer/Lösemittel Systemen ein reversibler Gel-Sol-Übergang [7-11].
|
|
|
|
|
|
|
|
|
Struktur-merkmale |
PUR-Typen |
Anwendungsgebiete |
|
linear |
Thermoplaste Elastomere PUR-Kautschuk Gießelastomere |
|
|
leicht verzweigt |
flexible und semiflexible Schäume |
|
|
verzweigt |
Beschichtungen |
|
|
stark verzweigt |
feste Schäume (Hartschaumstoffe) |
|
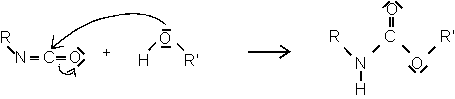 Isocyanat Alkohol Urethan
Aufgrund ihrer hohen Reaktivität gehen die Isocyanate noch eine Reihe von weiteren Reaktionen ein, die allgemein in zwei Klassen unterteilt werden können [38].
Isocyanat Alkohol Urethan
Aufgrund ihrer hohen Reaktivität gehen die Isocyanate noch eine Reihe von weiteren Reaktionen ein, die allgemein in zwei Klassen unterteilt werden können [38].
|
Reaktion der Isocyanate mit |
Produkte |
|
|
primären Aminen |
disubstiuierter Harnstoff |
|
|
sekundären Aminen |
trisubstituierter Harnstoff |
|
|
Carbonsäuren |
Amide |
|
|
Wasser |
instabile Carbaminsäure; zerfällt in Kohlendioxid und Amin |
|
|
Urethanen |
Allophanate; bei terminalen NCO-Gruppen vernetzte Strukturen |
|
|
Harnstoff |
Biuret; vernetzte Strukturen |
|
|
Amiden |
Acylharnstoffe |
|
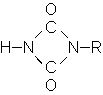
| Dimerisierung: |
|
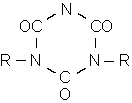
Trimerisierung: |
|
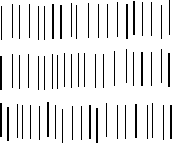 |
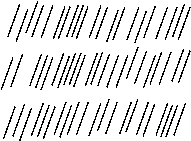 |
| Smektisch A: Schichten parallel angeordneter, stäbchenförmiger Mesogene; die Stäbchenachsen stehen senkrecht zu den Schichtebenen [42]. | Smektisch C: ähnlich der Struktur Smektisch A; die Molekülachsen sind zu den Schichtebenen geneigt [42]. |
 Abb.(4.2): Strukturmodell der nematischen Phase: Parallelorientierung der Moleküllängsachsen, die Molekülschwerpunkte sind statistisch verteilt.
Eine ideale Parallelordnung aller Moleküle einer nematischen Phase kann nur unter Einwirkung zusätzlicher Kräfte, wie zum Beispiel magnetischer oder elektrischer Felder, erreicht werden. In realen nematischen Flüssigkeiten ändert sich dagegen die Vorzugsrichtung kontinuierlich. Im Gegensatz zu den cholesterischen flüssigen Kristallen ist der Winkel, um den sich die Vorzugsrichtung von Ort zu Ort ändert, nicht definiert. Das Ausmaß der Parallelorientierung wird durch den Ordnungsgrad S charakterisiert, einem Zahlenwert zwischen Null für die ungeordnete isotrope und Eins für die ideal geordnete nematische Phase.
Abb.(4.2): Strukturmodell der nematischen Phase: Parallelorientierung der Moleküllängsachsen, die Molekülschwerpunkte sind statistisch verteilt.
Eine ideale Parallelordnung aller Moleküle einer nematischen Phase kann nur unter Einwirkung zusätzlicher Kräfte, wie zum Beispiel magnetischer oder elektrischer Felder, erreicht werden. In realen nematischen Flüssigkeiten ändert sich dagegen die Vorzugsrichtung kontinuierlich. Im Gegensatz zu den cholesterischen flüssigen Kristallen ist der Winkel, um den sich die Vorzugsrichtung von Ort zu Ort ändert, nicht definiert. Das Ausmaß der Parallelorientierung wird durch den Ordnungsgrad S charakterisiert, einem Zahlenwert zwischen Null für die ungeordnete isotrope und Eins für die ideal geordnete nematische Phase.
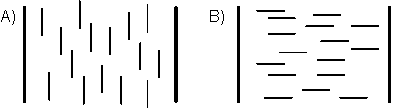 Abb.(4.3): Orientierung der Moleküle in dünnen nematischen Schichten [42]:
A) homöotrope oder senkrechte Orientierung
B) homogene oder parallele Orientierung
Einheitlich orientierte nematische Präparate (S=1) sind optisch und dielektrisch einachsig, wobei die Symmetrieachse parallel zur Molekülachse verläuft. Die Moleküllängsachse ist bei nematischen Flüssig-Kristallen zugleich die Richtung der größten Polarisierbarkeit des Moleküls. Man beobachtet stets Doppelbrechung.
In dünnen nematischen Schichten können sich die Moleküle nematischer Phasen mit ihren Längsachsen entweder parallel (homogene Orientierung) oder senkrecht (homöotrope Orientierung) zu den Grenzflächen einstellen. Die Anordnung der Moleküle an den Grenzschichten überträgt sich durch Dispersionswechselwirkungen auf die inneren Moleküle.
Abb.(4.3): Orientierung der Moleküle in dünnen nematischen Schichten [42]:
A) homöotrope oder senkrechte Orientierung
B) homogene oder parallele Orientierung
Einheitlich orientierte nematische Präparate (S=1) sind optisch und dielektrisch einachsig, wobei die Symmetrieachse parallel zur Molekülachse verläuft. Die Moleküllängsachse ist bei nematischen Flüssig-Kristallen zugleich die Richtung der größten Polarisierbarkeit des Moleküls. Man beobachtet stets Doppelbrechung.
In dünnen nematischen Schichten können sich die Moleküle nematischer Phasen mit ihren Längsachsen entweder parallel (homogene Orientierung) oder senkrecht (homöotrope Orientierung) zu den Grenzflächen einstellen. Die Anordnung der Moleküle an den Grenzschichten überträgt sich durch Dispersionswechselwirkungen auf die inneren Moleküle.
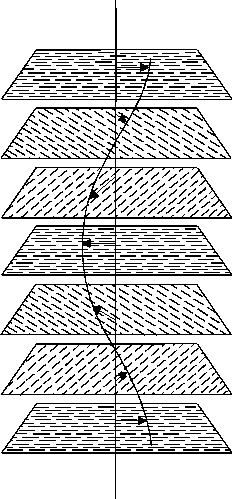 Abb.(4.5): Strukturmerkmal der cholesterischen Phase: Die Richtung der Moleküllängsachsen (Pfeile) ändert sich von Schicht zu Schicht um einen konstanten Winkel [42].
Zwischen der cholesterischen und nematischen Struktur besteht eine nahe Verwandtschaft. Sie wird besonders dadurch deutlich, daß durch Mischen von Cholesterinestern mit Rechtshelix- und Linkshelix-Anordnungen bei einem bestimmten Mischungsverhältnis und einer definierten Temperatur ein nematischer Flüssig-Kristall entsteht [47]. Bei dieser Temperatur heben sich die Verdrillungskräfte der Mischungskomponenten gegenseitig auf. Außerdem kann die cholesterische Struktur durch Lösen chiraler, optisch-aktiver Substanzen in nematischen Flüssigkeiten oder durch Spaltung nematischer Racemate erzeugt werden.
Mit dem helixartigen Aufbau der cholesterischen Phasen hängen die meisten ihrer optischen Eigenschaften zusammen. In diesem Zusammenhang sind ihre extrem hohe optische Rotation, der Zirkulardichroismus und die vom technischen Standpunkt wichtige selektive Reflexion des Spektralbereiches, dessen Wellenlänge der Ganghöhe der Helix entspricht, zu nennen [48]. Eine Änderung der Ganghöhe, etwa durch thermische, magnetische oder elektrische Energie, drückt sich in einer Änderung aller optischen Eigenschaften aus.
Abb.(4.5): Strukturmerkmal der cholesterischen Phase: Die Richtung der Moleküllängsachsen (Pfeile) ändert sich von Schicht zu Schicht um einen konstanten Winkel [42].
Zwischen der cholesterischen und nematischen Struktur besteht eine nahe Verwandtschaft. Sie wird besonders dadurch deutlich, daß durch Mischen von Cholesterinestern mit Rechtshelix- und Linkshelix-Anordnungen bei einem bestimmten Mischungsverhältnis und einer definierten Temperatur ein nematischer Flüssig-Kristall entsteht [47]. Bei dieser Temperatur heben sich die Verdrillungskräfte der Mischungskomponenten gegenseitig auf. Außerdem kann die cholesterische Struktur durch Lösen chiraler, optisch-aktiver Substanzen in nematischen Flüssigkeiten oder durch Spaltung nematischer Racemate erzeugt werden.
Mit dem helixartigen Aufbau der cholesterischen Phasen hängen die meisten ihrer optischen Eigenschaften zusammen. In diesem Zusammenhang sind ihre extrem hohe optische Rotation, der Zirkulardichroismus und die vom technischen Standpunkt wichtige selektive Reflexion des Spektralbereiches, dessen Wellenlänge der Ganghöhe der Helix entspricht, zu nennen [48]. Eine Änderung der Ganghöhe, etwa durch thermische, magnetische oder elektrische Energie, drückt sich in einer Änderung aller optischen Eigenschaften aus.